Более полувека ученые-биологи бились над проблемой предсказания трехмерной структуры белка по составляющим его аминокислотам. Хотя и решаемая, эта задача требовала огромных вычислительных ресурсов. Теперь же благодаря алгоритмам глубинного машинного обучения процесс, занимавший месяцы, может сократиться до пары недель или — на мощном оборудовании — нескольких часов.

Белковые молекулы — цепочки, составленные из аминокислот — представляют собой основу жизни и являются основным предметом изучения в молекулярной биологии, биохимии и многих областях медицины. Однако зная лишь формулу белка, предсказать варианты его взаимодействия с другими молекулами и клетками (а, значит, функции белка в организме) невозможно. Дело в том, что его свойства зависят от формы «укладки» полипептидной цепочки — а на эту форму влияет огромное число факторов. Например, гидрофобность, гидрофильность или электрический заряд составляющих белок аминокислот.
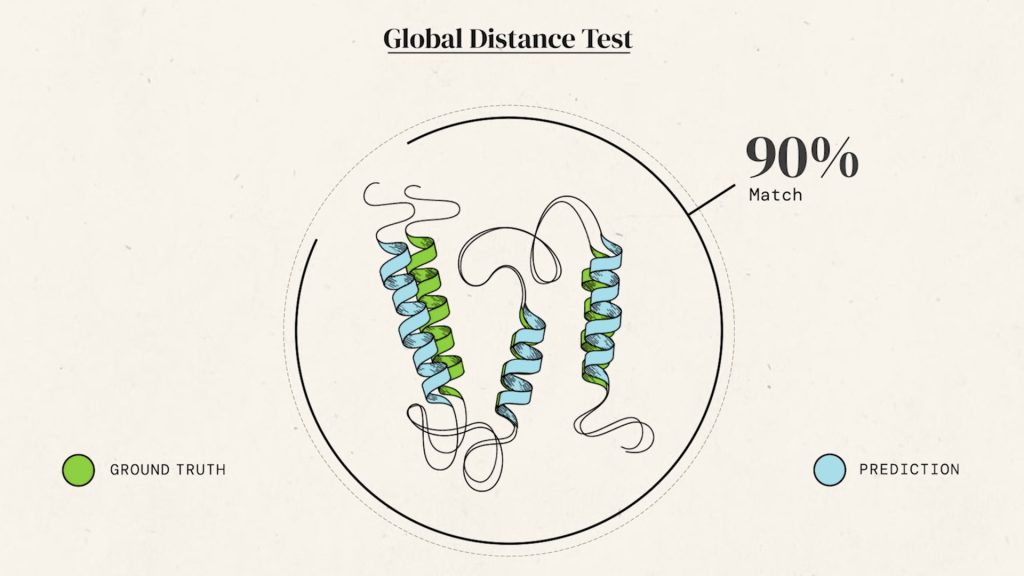
Решить так называемую задачу фолдинга («укладки») белка с достаточной точностью смогла последняя версия разработанного британской компанией DeepMind (принадлежит, наряду с Google, холдингу Alphabet) алгоритма AlphaFold. В компании накануне сообщили, что их разработку признали эффективной (точность предсказания — выше 90%) организаторы программы мониторинга исследований в области предсказания структуры белка (Critical Assessment of protein Structure Prediction, CASP).
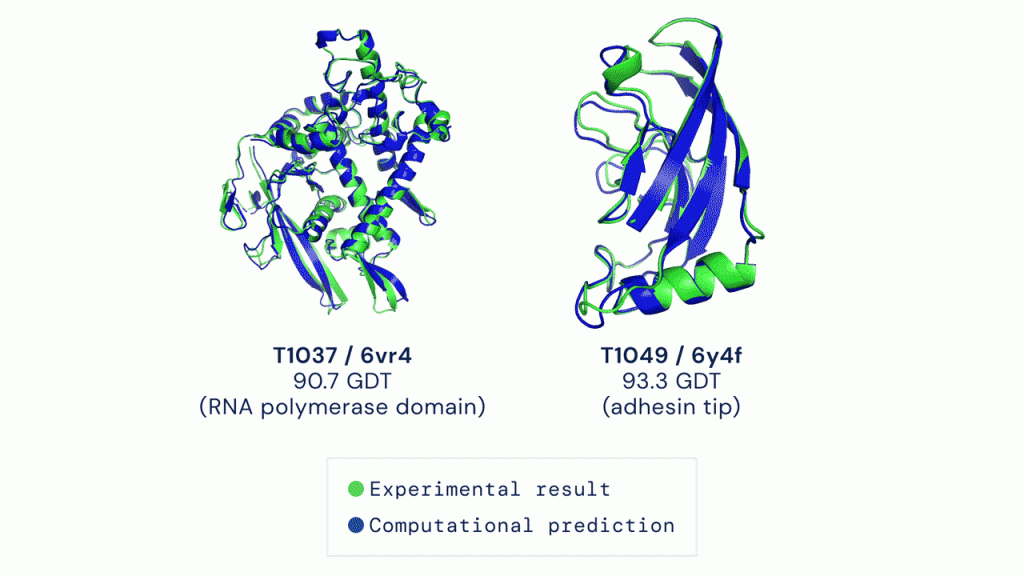
Не имея способа быстро предсказывать пространственную структуру белка, ученые прибегали к сложным и дорогостоящим техникам — от спектроскопии ядерного магнитного резонанса до рентгеноструктурного анализа. Однако все они так или иначе подразумевали поиск решения методом проб и ошибок. Новый вычислительный инструмент позволит, в частности, гораздо быстрее находить лекарства от многих болезней или создавать белки-ферменты, которые смогут эффективно перерабатывать загрязняющие окружающую среду вещества.
Первую версию AlphaFold DeepMind представила в 2018-м году — уже тогда это решение лидировало по эффективности среди аналогов в предыдущем обзоре CASP. С тех пор алгоритм, основанный на глубинном обучении, был усовершенствован с учетом последних открытий в области биологии, физики и искусственного интеллекта. Тренировали его на доступных данных о примерно 170 000 белковых структур, а также базах данных о белках с известной последовательностью аминокислот, но неизвестной структурой.